Supervised by: Merissa Hickman BSc (Hons). Merissa studied Biomedical Science at the University of Hull. She is a current postgraduate student at the University of Cambridge studying Genomic Medicine. She was awarded a Cambridge Trust Scholarship to complete her MPhil.
Abstract
Dihydropyrimidine dehydrogenase (DPD) testing measures the DPD enzyme activity, which is responsible for breaking down thymine and uracil as well as fluoropyrimidine (FP) based drugs used in solid cancer chemotherapies. This paper discusses two methods of DPD testing; phenotyping, genotyping and their alternatives, while also evaluating their applications and accessibility globally. The DPYD gene codes for the DPD enzyme. Mutations/variations in the DPYD gene affect the DPD enzyme activity, leading to insufficient DPD activity which causes FP-related toxicities. DPD testing by phenotyping is primarily done through blood tests, measuring the levels of exogenous metabolites (generally uracil/dihydrouracil). DPYD genotyping involves the analysis of the genome to screen for mutations/variants on the DPYD gene. Globally, both methods are recommended to be performed for maximum accuracy. The results of the tests allow for the FP-based treatment to be personalised depending on the DPD activity to minimise adverse effects. Furthermore, European countries strongly advise DPD screening prior to treatment. Conversely, DPD testing isn’t recommended in Asian countries as the incidence of DPD deficiency is estimated to be extremely minimal in Asian populations. Moreover, the emerging methods for DPD testing continue to be researched to improve the accuracy and applicability of the DPD testing. For example, the liquid chromatography-ultraviolet (UPLC-UV) method aims to decrease the duration of the procedure while increasing the accuracy by analysing the precise levels of exogenous metabolites.
1. Introduction
1.1 Fluoropyrimidines
Fluoropyrimidines (FP) are antimetabolite drugs used to eliminate cancer cells with chemotherapy (Thorn et al., 2011). FP treats solid tumours in the gastrointestinal, breast, head and neck (Clasen, O’Quinn & Carver, 2023). It is seen that using FP for chemotherapy treatment is often well tolerated among patients but comes with risk factors and side effects that can cause complications. These complications can develop due to an individual’s medical history, use of other drugs for medical or non-medical purposes and even age can play a role (Clasen, O’Quinn & Carver, 2023). Common side effects for FP toxicity are fatigue, nausea, diarrhoea, coronary vasospasm and arrhythmias (Clasen, O’Quinn & Carver, 2023). Examples of fluoropyrimidines drugs include 5-fluorouracil (5-FU), capecitabine, and tegafur. 5-FU is usually given intravenously and 80% of it is metabolised in the liver (Thorn et al., 2011). Furthermore, Capecitabine and tegafur are both prodrugs of 5-FU (Thorn et al., 2011).
1.2 What is DPD & DPD Deficiency?
DPD (dihydropyrimidine dehydrogenase) is the enzyme responsible for breaking down thymine and uracil, an important part of 5-FU and capecitabine (Cancer Research UK, 2023). Therefore, without sufficient amounts of DPD enzymes made by the liver, an individual may potentially end up with fatal side effects when using drugs such as 5-FU or capecitabine. DPD deficiency occurs when individuals have low or no levels of the DPD enzyme, usually due to genetic mutations on the DPYD gene (Cancer Research UK, 2023). Partial DPD deficiency is when an individual has low or very low levels of DPD but it is not usually shown through symptoms unless you get tested (Cancer Research UK, 2023). Complete DPD deficiency is rare, while low or partial deficiency are widely seen (Cancer Research UK, 2023). As stated earlier, an individual is not aware of their DPD deficiency unless they are using drugs like 5-FU since many doctors require testing before prescribing an individual with FP’s. Furthermore, complete deficiency can be seen in babies through symptoms such as a small head size, development issues and fits (Cancer Research UK, 2023). When anticancer drugs such as 5-FU are prescribed to patients with DPD deficiency and sufficient amounts of DPD enzymes are not produced, severe toxicity will be experienced by the patient. This kind of toxicity is due to a genetic variation in the DPYD gene. According to a research publication by Forike et al., approximately 10-40% patients suffer from severe toxicity (Martens. et al., 2020). In addition, 20-30 out of 100 people suffer from severe side effects from 5-FU and capecitabine, while less than 1% die from the treatment (Cancer Research UK, 2023).
1.3 DPD Testing
DPD testing identifies whether patients are at risk of toxicity due to DPD deficiency while informing doctors of the safe drug dose range for that specific individual, creating an effective personalised treatment plan. The two main types of testing for DPD deficiency are DPD phenotyping and DPYD genotyping. DPD phenotyping is the measurement of DPD enzyme activity in one’s body, while DPYD genotyping is predicting DPD activity based on genetic variants in the genome (Salama, Gallo-Hershberg and Forbes, 2023). In brief, DPD testing has the potential to decrease the risk of unwanted toxicity and side effects from the use of chemotherapy drugs, such as 5-FU and capecitabine, by providing doctors with the individual’s DPD activity inorder to prescribe safe dosage of FU’s.
1.4 Aims and Objectives
The aim of this paper is to explore testing methods, both established and recent, for genes and their variants as they impact the activity levels of the enzymes involved in breaking down fluoropyrimidines in patients being treated for cancer using FP-based drugs. This paper also aims to explore methods for directly measuring the enzyme activity levels themselves.
2. DPD Phenotyping
2.1 What is DPD phenotyping?
Out of the two main procedures for DPD enzyme testing (phenotyping and genotyping), phenotyping is a sought after approach recommended and implemented globally. Dihydropyrimidine dehydrogenase (DPD) phenotype testing aims to measure the DPD enzyme activity in the body through mechanical means, without the consideration of the DPYD gene (coding for the DPD enzyme) and the plausible mutations/variations on the patients’ genome. The objective of DPD phenotyping is to deduce DPD enzyme activity through the levels of endogenous metabolite (specifically uracil or dihydrouracil) of the DPD enzyme in blood, thus to create an optimal treatment plan for solid cancer patients involving fluoropyrimidines (FP), such as 5-fluorouracil (5-FU), in order to avoid fatal toxicity (Amstutz et al., 2017; Hodroj et al., 2021; Henricks et al., 2018; Robin et al., 2020).
DPD phenotype testing is branched under two main categories: direct and indirect phenotyping as seen in Figure 1 (Hodroj et al., 2021). Indirect DPD phenotyping is performed in two common methods: evaluating the dihydrouracil/uracil (UH2/U) ratio in the blood or assessing the uracilemia (uracil concentration present in blood) in the blood while direct DPD phenotyping involves the evaluation of peripheral mononuclear cell DPD activity (PBMC-DPD activity) (Hodroj et al., 2021; Paulsen et al., 2022). Indirect DPD phenotyping is more commonly implemented globally as there is more clinical data and research on their application as well as higher credibility in results compared to direct phenotyping procedures (Paulsen et al., 2022).
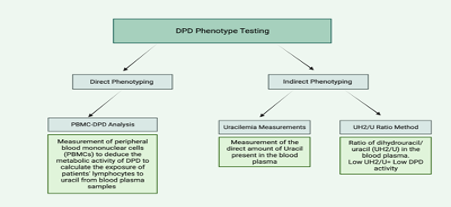
Figure 1. An Overview of the Main Types of DPD Phenotyping (Maillard et al., 2022; Paulsen et al., 2022; Meulendijks et al., 2017).
2.2 Uracilemia Measurements
The most common DPD phenotyping approach is the measurement of uracil, as it is metabolised by the DPD enzyme (Paulsen et al., 2022). Referred to as the measurement of uracilemia, this method measures the content of uracil present in blood plasma through two main approaches: blood sampling and urine sampling. The exact protocols for both blood and urine sampling are well described in a research publication by Jiang et al., though it is important to note that urine sampling is not preferred over blood sampling due to the effects of renal activity on results (Jiang et al., 2002; Royer et al., 2023). Table 1 showcases the uracilemia testing result indications according to the European Medicine Agency (EMA) (EMA, 2023).
| Blood Uracil Level | Indication |
| ≥ 16 ng/ml and < 150 ng/ml | Partial DPD deficiency |
| ≥ 150 ng/ml | Complete DPD deficiency |
Table 1. Uracilemia Testing Results Indications (EMA, 2023)
Despite being the most sought for method of phenotyping, uracilemia measurements come with various disadvantages. Firstly, there is a clear lack in literature focused on the clinical trials of uracilemia measurements, which means that the supporting verifications for the current threshold values of uracil concentration proposed by EMA is sparse (Paulsen et al., 2022). Additionally DPD activity alters greatly with exogenous factors such as food intake, circadian variation and preanalytical conditions (Jiang et al., 2002; Paulsen et al., 2022). Moreover, as the results of phenotyping are quantitative values, they’re uninterpretable without a robust clinical cut-off value to be assigned to patients to be led to specific dosing categories, which is caused by a lack of clinical research and continuous standardised testing between laboratories (Paulsen et al., 2022).
The most prominent reason for phenotyping anomalies are caused by renal impairment, which can lead to false-positive uracilemia results. In a study conducted by Royer et al., it was observed that there was an increased uracilemia as renal function decreased (Royer et al., 2023). Furthermore, they concluded that 86.4% of the patients in the clinical trials were sampled as DPD deficient before dialysis, compared to 13.7% after dialysis. Thus, it’s recommended for patients with a history of renal impairment to receive DPD phenotyping after dialysis to avoid false-positive results. Also, the use of 5-FU therapeutic drug monitoring is advised to create a personalised dosage, as well as frequent phenotyping check-ups while on chemotherapy (Royer et al., 2023).
Despite these disadvantages, uracilemia measurements continue to be the preferred line of testing for DPD deficiency due to being a minimal invasive procedure without the requirement of special equipment, and its ability to deflect current DPD activity while also detecting patients with rare DPYD variants (Paulsen et al., 2022).
2.3 UH2/U Ratio Method
The alternative approach to indirect DPD phenotyping is checking the dihydrouracil/uracil (UH2/U) ratio. The DPD enzyme converts uracil into dihydrouracil, thus the UH2/U ratio becomes an indirect method of measuring DPD activity (Meulendijks et al., 2017). Similar to uracilemia measurements, the methodology of measuring the UH2/U ratio follows a similar procedure that is found in the study by Robin et al. (Robin et al., 2020). Moreover, the limit of quantification was set at 5 and 10 ng/ml for U and UH2 respectively by the EMA (Kristensen, Pedersen, Mejer, 2010). In a study conducted by Gamelin et al., patients with a UH2/U ratio of <1.8 were observed to have adverse effects to 5-FU, thus 1.8 was identified as the border zone (Gamelin et al., 1999). The indications of UH2/U values are showcased in Table 2.
| UH2/U Ratio | Indication |
| High (>1.8) | Sufficient DPD activity |
| Low (<1.8) | Insufficient DPD activity |
Table 2. UH2/U Ratio Indications (Paulsen et al., 2022; Gamelin et al., 1999)
A research publication by Gaible et al. suggested that the ratio method’s advantage against uracilemia measurements was that it is a more reliable approach to phenotyping for patients with renal impairments (Gaible et al., 2021). An adverse argument was made by Royer et al. who observed in their clinical trials that dihydrouracil was impacted neither by renal impairment nor by dialysis (Royer et al., 2023).
Moreover, just like uracilemia measurements, UH2/U is more reliable due to the wider range of clinical data available and the affordability of the procedure. However, there is still a lack of clinical trials and standardised testing, making indirect phenotyping an area of research that must be further investigated. Furthermore, information about cost-effectiveness of DPD phenotyping is lacking, though UH2/U tests are performed at a price of $250.00 (Ockeloen et al., 2021; eviQ, 2023).
2.4 PBMC-DPD Testing
Referred to as the ‘gold-standard approach’, measuring DPD activity in peripheral blood mononuclear cells (PBMCs) is the direct phenotyping method (Maillard et al., 2022). DPD activity is found in most tissues but is highest in the liver tissue. PBMCs are used to monitor clinically DPD activity (Milano and Chamorey, 2002). In PBMC-DPD measuring, the metabolic activity of DPD is directly assessed by measuring the amounts of dihydrouracil, carbamyl-B-alanine, and B-alanine produced from exposure of patient lymphocytes to uracil from blood plasma samples (Hodroj et al., 2021). DPD activity measured in PBMC is correlated to FP systemic clearance (body’s ability to eliminate FP based drugs), though the correlation is weak. Nevertheless, patients with suspected or proven PBMC-DPD deficiency exhibit severe FP-related toxicities (Milano and Etienne, 1994). Moreover, DPD activity in tumour cells (in vitro and clinical studies) is significantly related to FP sensitivity: the lower the DPD activity, the greater the FP effectiveness. Hence, further prospective clinical studies will be required in order to confirm the correlation between PBMC-DPD activity and FP clearance, which becomes the method’s greatest disadvantage: lack of clinical research (Milano and Etienne, 1994).
Another limitation of PBMC-DPD testing is that in normal peripheral blood lymphocytes and intestinal mucosa, the catabolism of the uracil cannot be detected beyond dihydrouracil. This indicates a very low or a lack of dihydropyrimidinase activity (the enzyme that metabolises dihydrouracil), which creates potential false-positive results for DPD deficiency (Lu, Zhang and Diasio,1993; Gamelin et al., 1997). Moreover, in the limited number of clinical trials conducted, the correlation between PBMC-DPD activity and FP clearance can be considered weak, determining that PBMC–DPD is not sufficient to accurately predict FP clearance (Milano and Chamorey, 2002). PBMC-DPD testing is also not suitable for daily care due to the fact that it is affected by exogenous factors, such as circadian rhymes and food intake (Maillard et al., 2022). Moreover, PBMC-DPD testing is considered a labour-intensive method for which special equipment is needed, which is not available in every hospital so it is difficult to implement this method on a large scale (Ockeloen et al., 2021).
2.5 Applications
DPD phenotyping is recommended on a large scale globally, however significant changes in policies can be seen due to geographical changes. France has been the most prominent country in terms of their stance on DPD testing. Since September 2018, the French guidelines have recommended DPD deficiency screening by plasmatic uracil quantification before any FP-based chemotherapy prescription. Later in December 2018, prior to the initiation of treatment with fluoropyrimidines, a search for DPD deficiency was made mandatory by determining the uracilemia measurements (Gaible et al., 202; Laures et al., 2022). French guidelines recommend uracilemia measurements as the optimal phenotyping procedure due to it being the most reliable, however phenotyping by measuring plasma the UH2/U ratio has also been approved by the French Health Authorities (Haute Autorité de Santé, 2018). This is so due to the indirect phenotyping being a minimally invasive procedure while being the most reliable method due to having the most amount of clinical research. So far French guidelines have made it clear that PBMC-DPD testing is not their preferred approach to DPD phenotyping, presumably due to lack of reliable evidence for FP clearance and its need for special/expensive equipment as a way of creating financial burden on patients (Ockeloen et al., 2021).
Similarly, Since April 2020, the European Medicines Agency (EMA) recommends DPD testing before any FP-based treatment, however it is not mandatory. EMA recommends both direct and indirect method of phenotyping, though they account uracilemia measurements as the optimal testing procedure (Hodroj et al., 2021). More specifically, in Belgium, phenotyping that is based on plasma uracil levels is also recommended due to the same reason as the French policies (Laures et al., 2022).
Despite the similarities in recommended phenotyping approaches in Europe, this is not the case in Asia due to a primary reason; incidence of DPD deficiency is estimated to be extremely minimal in Asian populations compared with non-Asian populations (Yoshino et al. 2021). Thus instead of DPD testing prior to the FP-based treatments to personalise the dosages; testing is only done/recommended if the patient experiences severe FP-related adverse effects. This makes DPD phenotyping an uncommon practice in Asian countries in comparison to Europe.
Overall, DPD phenotyping is a sought after option, especially in Europe, due to being an accurate and minimally invasive procedure for patients. Nevertheless, all forms of DPD phenotyping require more clinical trials to further understand and optimise FP-based treatments for solid cancer patients.
3. DPYD Genotyping
3.1 What is genotyping?
Genotyping is a process which tells us a person’s DNA sequence at a specific position in the genome and is determined by the combinations of alleles inherited from both parents (Kockum, Huang & Stridh, 2023). The first step to genotyping is to have purified DNA. This can be done through the extracting of chromosomal DNA from the cellular nuclei, then removing proteins and cell debris which results in a purified DNA Sample (Kockum, Huang & Stridh, 2023). Detergent and mechanical shearing can be used for the extracting of chromosomal DNA (Kockum, Huang & Stridh, 2023). One can obtain DNA through blood, saliva, or paraffin-embedded biopsy tissues (Kockum, Huang & Stridh, 2023). An organic extraction of genomic DNA can be done by simply adding phenol/chloroform (Kockum, Huang & Stridh, 2023). This separates the DNA and proteins into different organic phases and requires little equipment, hence it is cheaper (Kockum, Huang & Stridh, 2023). Although there are more expensive ways to isolate and purify DNA for genotyping, commercial kits are the faster and easier method (Kockum, Huang & Stridh, 2023). Once you have a purified sample of DNA, you have the ability to check for genetic variants, which is crucial medical data as it helps doctors to make predictions about an individual’s body for more personalised treatments.
DPYD genotyping is one of the most widely used methods to assess DPD activity in an individual. Genotyping is also the more clinically accepted method for testing DPD deficiency as it refers to the use of DNA to determine the DPYD variants present in one’s body (Innocenti et al., 2020). Usually patients are able to tolerate fluoropyrimidines without any adverse effects but studies show that a third of patients often end up developing toxicities caused by the treatment (Salama, Gallo-Hershberg & Forbes, 2023). The adverse side effects of fluoropyrimidines are due to the genetic difference in metabolism of 5-FU. Hence, doctors need to review an individual’s genetic variations before prescribing certain amounts of fluoropyrimidines. Thankfully, several genetic variants, which impact the enzymatic activity of a DPD and increase the risk of toxicity, were identified (Table 3). Reduced DPD activity is approximately 3-5% in the general population, while in caucasians it has been estimated to be 7% (Salama, Gallo-Hershberg & Forbes, 2023). This statistic shows that race and ethnicity are factors of genetic variations.
| DPYD Variants | Functional Status |
| Wild-type: (DPYD*5) (DPYD*9A) | Normal activity |
| (c.2846A>A) | Decreased activity |
| (c.1236G>A) (c.1129-5923C>G) | Decreased activity |
| (DPYD*2A) | NO activity |
| (DPYD*13) | NO activity |
Table 3 – DPYD variants that are known to reduce DPD activity (Salama, Gallo-Hershberg & Forbes, 2023).
3.2 Applications & Advantages
The Netherland’s National guideline for colon carcinoma recommends DPYD genotyping before treatment with FP (Martens, 2020). Studies in the Netherland’s highlighted the cost saving associated with testing DPYD variants, one of many advantages of genotyping. In Ontario specifically, an approximate saving of $714,963 was reported for the next 5 years if DPYD genotyping is implemented for patients being treated by FP (Salama, Gallo-Hershberg & Forbes, 2023). In addition, the European Society for Medical Oncology also recommended genetic testing through a document in 2020 (Innocenti et al, 2020). Also, in the UK, the National Health Service recommended using DPYD genotyping when screening for DPD deficiency. Furthermore, DPYD genotyping is a simple and well-known technique, which usually has little effect on an individual’s day-to-day life. In brief, DPYD variant testing seems to be recommended around the world for being a simple procedure while being able to detect whether an individual is complete, partial or has no deficiency.
In addition, it can be seen that if DPYD genotyping can decrease the incidence of adverse effects and severe toxicity, it is also expected that there will be a decrease in the cost of healthcare. This point was investigated by Deenen et al. and Mericer et al. A the study by Deenen et al. showed that the average total treatment cost per patient were lower for upfront DPYD*2A-screening, DPD variant, than for non-screening (Deenen et al., 2016). A study by Mercier et al. resulted in the conclusions that dose individualization based on DPYD genotype or DPD phenotype is cost-saving (Mercier et al., 2009).
3.3 Limitations
One of the many limitations to DPYD genotyping is that it can only examine known DPYD variants as there are still many rare and undiscovered variants which affect DPD. This makes it hard to detect other DPD deficiencies that cannot be explained by genetic variants, hence why phenotyping is additionally implemented. Therefore, by discovering as well as researching these new and rare variants, it will strengthen the predictive power of FP toxicity from DPYD genotype. By combining DPYD genotyping and DPD phenotyping, the results are more likely to be accurate compared to using only one method (Paulsen et al, 2022). A reason for this method not being implemented across the world is because of how expensive these tests can be. Another limitation to DPD testing is that reduced DPD activity only affects about 3-5% of the general population and in caucasians, the prevalence of these specific variants is estimated to be 7% (Salama, Gallo-Hershberg & Forbes, 2023). However, another study suggested the inclusion of the (c.496A>G) variant in routine DPD testing among South Asian populations would be beneficial as this variant is more commonly seen among the South Asian population (Hariprakash et al., 2018).This indicates that there are distinctions among different racial and ethnic groups. Therefore, the discovery of new variants are needed as well as variants affecting specific groups, so tests can be more inclusive to the general population. Lastly, DPYD genotyping is currently limited due to the cost and resources required to sequence all clinically relevant DPYD variants. This shows that there are still barriers to DPYD genotyping that urgently need to be overcome.
4. Future of DPD Testing
4.1 New discoveries within DPYD testing
With the development of improved and cheaper genome sequencing technology, new clinically relevant genes are being discovered at a rapid pace. Continued DPYD exome sequencing is finding new and rare DPYD variants, such as the novel deleterious mutations c.2087>G and c.2324T>G, and a novel variant p.L775W indicative of DPD deficiency (García-González et al., 2020). Additionally, the discovery of new clinically relevant DPYD variants is beginning to extend to other non-European ancestry, increasing multi-ethnic applicability (da Rocha et al., 2021). DPYD-Y186C variant was found to be unique to African ancestry and was associated with a 46% decreased DPD activity, and homozygous carriers of C29R showing a 27% increased DPD activity (Offer et al., 2013). Haplotype analysis is also increasing. A Multi-SNP analysis indicated a correlation between three-SNP haplotypes (Hap7) involving rs1801160, rs1801265 and rs2297595 and severe FU toxicity comparable to the splice site variant rs3918290, indicating haplotype analysis may also be a strong indicator of 5-FU toxicity (Gentile et al., 2015). A retrospective study done by T. K. Froehlich et al. found further clinically relevant DPYD haplotypes. c.1129–5923C>G/hapB3 was associated with severe toxicity , with 55% of patients requiring dose interventions (Froehlich et al., 2014). Epigenetic testing of the DPYD gene has also shown some predictive powers, with methylation of DPYD promoter regions being detected in DPD deficient patients (Ezzeldin et al., 2005). Additionally, methylation of CpG islands in the 5’ regulatory region of the DPYD gene promoter is also reported to potentially inhibit the binding of transcriptional factors or impact chromatin structure stability and thus directly inhibiting transcription. However, certain studies show contradictory findings with weak methylation patterns being detected in DPD deficient patients, suggesting that methylation patterns are only a slight and weak predictor of FP toxicity (Amstutz et al., 2008). Finally, Epigenetic testing of the DPYD gene has also shown some predictive powers (Ezzeldin et al., 2005).
4.2 Emerging methods for DPD testing and its alternatives
The coding of the DPD enzyme is not only attributed by the DNA DPYD gene, but also mRNA transcripts that are ultimately translated to synthesise DPD. Increased accumulation of RNA induced silencing complex (RISC) proteins have been found in cells with highly homologous microRNAs miR-27a and miR-27b overexpression on DPYD mRNA that caused repression of DPD expression via two conserved recognition sites in the DPYD gene (Offer et al., 2014). This resulted in a significantly lower half maximal inhibitory concentration value of 4.4 μM versus the normal 14.3 μM. This was further validated by a mouse model study, which showed an inverse correlation with DPD activity and the expression levels of miR-27a and miR-27b. A common variant of the hairpin loop region of hsa-mir-27a (rs895819) was also associated with elevated miR-27a expression and a reduced DPD enzyme activity in both a transgenic overexpression model and a patient study. Offer et al. (2014) also associated the elevated presence of AGO proteins, which are the catalytic components of the RISC, with DPYD mRNA in cells with miR-27a and miR-27b overexpression, serving as another biomarker for DPD activity. Another study by Genet et al. conducted a deep intronic gene analysis, which showed a novel mutation c.1129–5923C>G, identified by creating a cryptic splice donor site in addition to a deleterious c.1129–5923C>G mutation associated with intronic polymorphisms (van Kuilenburg et al., 2010). Genomic DNA from EDTA-blood was isolated for MLPA testing using DPYD probes, revealing associations with toxicity. Array CGH with Agilent slides assessed genomic variation. Sequence analysis of cDNA and gDNA for DPYD was performed via PCR amplification. Quantitative PCR analysis studied wild-type DPD cDNA and variants. Homology modelling of human DPD used a pig DPD crystal structure as a basis.
Whilst extensions from DPYD gene testing on assessing DPD activity are rising, as mentioned previously, genes coding for relevant FP metabolism pathway enzymes and proteins are slowly being exposed and researched. The gene TYMS codes for the enzyme thymidylate synthase (TS), responsible for the methylation of deoxyuridylate into deoxythymidylate, integral to FP metabolism and can be polymorphic, leading to altered TS activity and causing FP toxicity. A meta-analysis by B. A. Jennings et al. found that genetic variants of TYMS with 5’ UTR tandem repeats (rs45445694) tend to decrease TS activity, with the 2R/2R variant expressing the lowest activity (Jennings et al., 2012). However, this was associated with a small, but statistically significant clinical benefit, suggesting that TYMS can predict DPD activity to a small extent. MTHFR genetic variations that code for methylenetetrahydrofolate reductase, which is also involved in the FP metabolism pathway, showed contradictory findings (Afzal et al., 2011; Jennings et al., 2012). Its predictive power for FP toxicity was slight and statistically insignificant. A recent study by Rosmarin et al. made significant progress to the TYMS-DPD activity. The SNP rs2612091 is a common variant in the downstream of TYMS and intronic to ENOSF1, which explains TYMS genetic variations such as 5’ VNTR 2R/3R and 3’ UTR 6 bp ins-del. However, interestingly, the analysis of public mRNA expression demonstrated rs2612091 to be associated with ENOSF1 expression and not with TYMS expression , hence implying that while ENOSF1 and TYMS transcripts are overlapping, 5-FU “toxicity SNP does not appear to act through antisense-mediated down-regulation of TYMS mRNA (Rosmarin et al, ). Furthermore, they have found additional common DPYD variants with capecitabine toxicity: A-allele of SNP rs12132152 and rs76387818, a variant in the region flanking this tag SNP, with rs76387818 displaying a slightly higher severity of toxicity.
To improve the accuracy of prescriptions, in-silico analysis (computer-based analysis) genetic variants have been developed. In-silico analysis involves computerised calculations and simulations on the three-dimensional structure of enzymes and thus can accurately predict and model their activity (Yokoi et al., 2020). This technology would only become more accurate and quicker in the future. Furthermore, in-silico analysis can more accurately predict enzymatic activity and thus drug toxicity as the digital simulation is fully controlled.
While the previously mentioned alternative methods for DPYD testing are very accurate and holistic in terms of the FP metabolic pathway, due to the escalating number of patients requiring analysis, there was an urgent need to develop straightforward, rapid, and cost-effective methodologies suitable for routine screening purposes that the emerging genotyping methods lacked. To address this critical need, a novel and robust ultra performance liquid chromatography-ultraviolet (UPLC-UV) method was devised (Marin et al., 2020). Marin et al. developed this method through the preparation of stock solutions of U, UH2, and an internal standard 5-FU in ultrapure water. Working solutions were created by diluting stock solutions, and calibration standards and quality control samples were prepared by adding working solutions to bovine serum albumin (BSA). Chromatographic separation was performed using an H-CLASS Acquity UPLC system with an Acquity UPLC HSST3 analytical column. Sample preparation included transferring plasma to a hemolysis tube, followed by solid-phase extraction (SPE) using CX100-Interchim cartridges. This method enables the detection of uracil (U) and dihydrouracil (UH2) in plasma samples. Notably, this method achieved a significant reduction in analytical run-time, now condensed to 12 minutes, making it well-suited for large-scale initial screening efforts. While some HPLC methods significantly reduced their analytical run-time to 15 minutes, they are not suitable for extensive screening efforts due to their necessitating a significant plasma volume. The UPLC-UV method effectively differentiates patients with DPD deficiency from those without as it succeeds in distinguishing non-deficient patients (with U levels below 16 ng/ml) from deficient patients (with U levels exceeding 16 ng/ml). In a practical application of the UPLC-UV method, a month-long screening initiative was undertaken, involving a total of 263 blood samples collected from various institutions. The method effectively pinpointed patients exhibiting varying degrees of DPD deficiency. Impressively, 10.7% of the screened patients exhibited indications of deficiency that warranted dosage modifications. The development of the UPLC-UV method for DPD testing addresses a critical necessity in clinical oncology by providing a rapid, uncomplicated, and economical approach to ascertain DPD levels (Marin et al., 2020). However, while UPLC-UV is time and cost-effective, most UPLC-based methods for U and UH2 analysis are coupled with MS/MS detection, limiting the number of laboratories that are able to implement the method at low cost using basic double-wavelength UV detection. Additionally, the stability of blood samples are also quite sensitive which can cause difficulties in sample storage.
While DPD deficiency is responsible for roughly 43% of severe grade 3-4 side effects linked to 5-FU, a portion of patients continue to exhibit toxicity associated with the drug despite having normal DPD enzyme activity. This hints at potential deficiencies in the other enzymes involved in the 5-FU drug metabolism pathway. Dihydropyrimidinase (DHP), encoded by the DPYS gene, and/or beta-ureidopropionase (β-UP), encoded by the UPB1 gene, are two of such enzymes. The final enzyme involved in the breakdown of 5-FU is β-UP. β-UP catalyses the last step in the breakdown of fluoropyrimidines (Hishinuma et al., 2020). However, despite this enzymatic role, the connection between β-UP activity and FP-related toxicity remains unclear (Kuilenburg et. al., 2003). Similarly to DHP deficiency, the scarcity of β-UP is frequently observed in East Asian populations, including Japan and China. The initial step of 5-FU breakdown involves the DPD enzyme catalysing the conversion of 5-FU into FUH2. Following this, FUH2 can undergo further decomposition facilitated by DHP and β-UP, resulting in the production of fluoro-β-ureidopropionate (FUPA) and, subsequently, fluoro-β-alanine (FBAL). In a study that examined the DHP gene of 23 tumour patients with normal DPD activity, a heterozygous 833G>A mutation was identified in the DHP gene of one individual. The resulting mutant DHP protein exhibited complete inactivity. Notably, complete DHP deficiency is rare, with only nine cases documented as of 2003. However, in Japan, the prevalence of DHP deficiency has been approximated at 1 in 10,000 individuals, which suggests that DHP deficiency might be less uncommon than previously thought (Kuilenburg et al., 2003). While DHP deficiencies are not often documented among Caucasians, they exhibit a notable prevalence within Asian populations. These findings suggest that DHP deficiency can contribute to severe drug toxicity in patients undergoing 5-FU treatment, even if their DPD activity is normal. However, further investigation is required to thoroughly understand the relationship between DHP deficiency and 5-FU toxicity (Kuilenburg et al., 2003).
4.3 Future of DPD testing
With the advent and continuous improvement of next generation sequencing as well as an increase in novel clinically relevant DPYD gene variant discovery rate, most genetic variants of DPYD that are associated with DPYD toxicity should be encoded. This should ideally carry to other ancestries other than the European ancestry, though this is already in motion with more Asian and African DPYD gene variants such as rs115232898-C and Y186C being sequenced (Yap et al., 2017; Cunha-Junior et al., 2019; da Rocha et al., 2021). However, despite the increasing technology, a dosing algorithm must be developed to incorporate these DPYD genetic variants clinically to reduce FP toxicity. For this to occur, more clinical studies focusing on the toxicity effects of DPYD variants should proceed, with multiple studies conducted for each clinically relevant DPYD variant to reduce error-induced inaccurate results and verify existing toxicity risks. However, due to the decreasing costs and increasing speed of genome sequencing, a wider view of DPD activity should be the final target. As the purpose of DPYD testing is to assess patient risks of FP drug toxicity, a systematic genotyping/phenotyping system should be implemented to test the whole range of FP drug metabolism. For example, testing for target receptors and transport proteins with the current DPYD testing system included as a subset, whereby patients will have the relevant areas of their genome sequences prior to FP chemotherapy. This will drastically increase FP toxicity predictions and decrease FP toxicity. However, for this to occur, an extremely vast and robust bioinformatic and genomic database must be in place with rapid testing technology and an infrastructure to accommodate all of this smoothly. Thanks to the rapidly developing computer/database storage and genotyping testing technology, the former would not be a significant issue in the future. A complex and accurate algorithm construction through rigorous clinical studies and testing should then be top point, with a computerised system generating optimal treatment plans based on the genotype data available.
As a last note, the development of DPYD testing and its alternatives are to assess FP based chemotherapy toxicity in cancer patients. However, it does raise the question of whether FP toxicity, particularly the widely used 5-FU, could be avoided. A Japanese drug research team developed a FP based drug with similar effectiveness to 5-FU in patients with colorectal cancer while exhibiting less toxicity (Jiezhong et al., 2016). This drug, TAS-102, passed its clinical trials recently and further developments of this drug will likely ensue in the following years, which could drastically reduce FP toxicity. However as of now, TAS-102 has shown a lower overall survival period compared to 5-FU and other FP drugs, but synergizing agents are being investigated as well as further developments to the drug to improve its efficacy. Additionally, TAS-102 still has limited clinical data due to the drug being fairly new and not yet widely implemented. Thus, further studies must ensue to thoroughly assess toxicity or other side effects to compare with FPs. Ultimately, this is the final goal, as toxicity risk pre-emptive genetic tests would not have to occur if the drug presented little toxicity in the first place.
5. Conclusion
In the advent of personalised medicine, genotyping and phenotyping technology of DPYD provided clinical insights to the toxicity of chemotherapeutic agents, particularly fluoropyrimidines, allowing for personalised cancer therapy to arise. Indirect and direct methods of phenotyping, analysing DPD activity directly and genotyping methods, and analysis of the activity of DPD through the DPYD gene, can accurately predict the activity of DPD and thus the patient’s toxicity susceptibility to FP based chemotherapy. As of currently, these testing methods are imperfect, with limited ethnicity scope, undefined dosage guidelines based on genetic data, and lengthy testing durations. However, improvements are on the horizon. With the advent of advanced genome sequencing methods, an increasing number of clinically relevant DPYD gene variants are being discovered. Additionally, translation level screening methods are also developing, with mRNA analysis showing successful DPD activity predictions. A deeper understanding of the FP metabolic pathway has opened up further screening methods for clinically relevant genes involved in the pathway. The most beneficial usage of these screening methods would be in convergence, with both phenotyping and genotyping testing used to determine FP toxicity, boosting their toxicity predictive powers. However, this will likely be costly and logistically demanding, hence a robust and extensive sequencing, screening, analysis, and consulting system must be developed with advanced bioinformatic infrastructure to hold the vast amount of clinical genomic information. Our final recommendation would optimally be to integrate both types of methods prior to FP-based chemotherapy to improve testing accuracy and patient therapy outcomes.
Bibliography
Amstutz, U. et al (2008). Hypermethylation of the DPYD promoter region is not a major predictor of severe toxicity in 5-fluorouracil based chemotherapy. Journal of Experimental & Clinical Cancer Research, 27(1). doi:10.1186/1756-9966-27-54.
Amstutz, U. et al. (2017). Clinical pharmacogenetics implementation consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clinical Pharmacology & Therapeutics, 103(2), pp. 210–216. doi:10.1002/cpt.911.
Amstutz, U., Henricks, L.M., Offer, S.M., Barbarino, J., Schellens, J.H.M., Swen, J.J., Klein, T.E., McLeod, H.L., Caudle, K.E., Diasio, R.B. and Schwab, M. (2017). Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clinical Pharmacology & Therapeutics, 103(2), pp.210–216. doi:https://doi.org/10.1002/cpt.911.
Campbell, N. (2022). Dihydropyrimidine dehydrogenase (DPD) deficiency, NHS choices. Available at: https://www.genomicseducation.hee.nhs.uk/genotes/knowledge-hub/dihydropyrimidine-dehydrogenase-dpd-deficiency/ (Accessed: 23 August 2023).
Chen, Jiezhong et al. (2016). “TAS-102 an Emerging Oral Fluoropyrimidine.” Anticancer research.. 36(1), pp.21-6.
Cancer Research UK. (2023). DPD deficiency. https://www.cancerresearchuk.org/about-cancer/treatment/chemotherapy/side-effects/dpd-deficiency [Accessed 17th August 2023].
Chemistry LibreTexts. (2023). Internal Standards and LOD Available at: Internal Standards and LOD – Chemistry LibreTexts [Accessed 18 Aug. 2023].
Clasen, S., O’Quinn, R. & Carver, J. R (2023). Fluoropyrimidine Chemotherapy.https://www.cardiosmart.org/topics/cancer-treatment-and-your-heart/fluoropyrimidine-chemotherapy [Accessed 23th August 2023]
Cunha-Junior, G.F. et al (2019). Prevalence of the DPYD variant (Y186C) in Brazilian individuals of African ancestry. Cancer Chemotherapy and Pharmacology, 84(6), pp. 1359–1363. doi:10.1007/s00280-019-03974-4.
da Rocha, J.E., Lombard, Z. and Ramsay, M (2021). Potential impact of DPYD variation on fluoropyrimidine drug response in sub-Saharan African populations. Frontiers in Genetics, 12. doi:10.3389/fgene.2021.626954.
Deenen, M. J., Meulendijks, D., Cats, A., Sechterberger, M. K., & Severens, J. L., Boot, H., Smits, P. H., Rosing, H., Mandigers, C. M. P. W., Soesan, M., Beijnen, J. H. & Schellens, J. H. M. (2016). Upfront genotyping of DPYD*2A to individualise fluoropyrimidine therapy: a safety and cost analysis. Journal of Clinical Oncology. 34(3), 227-34. https://dspace.library.uu.nl/handle/1874/345672
EMA. (2023). 5-Fluorouracil (i.v.), capecitabine and tegafur containing products: Pre-treatment testing to identify DPD-deficient patients at increased risk of severe toxicity. Available at: 5-Fluorouraci capecitabine and tegafur containing products – DHPC and communication plan [Accessed 18 Aug. 2023].
eviQ. (2023). 1744-Dihydropyrimidine dehydrogenase (DPD) enzyme deficiency. eviQ. [online] Available at: 1744-Dihydropyrimidine dehydrogenase (DPD) enzyme deficiency | eviQ . [Accessed 18 Aug. 2023]
Ezzeldin, H.H. et al. (2005). Methylation of the dpyd promoter: An alternative mechanism for dihydropyrimidine dehydrogenase deficiency in cancer patients. Clinical Cancer Research, 11(24), pp. 8699–8705. doi:10.1158/1078-0432.ccr-05-1520.
Froehlich, T.K. et al. (2014). Clinical importance of risk variants in the dihydropyrimidine dehydrogenase gene for the prediction of early-onset fluoropyrimidine toxicity. International Journal of Cancer [Preprint]. doi:10.1002/ijc.29025.
Gaible, C., Narjoz, C., Loriot, M.-A., Roueff, S. and Pallet, N. (2021). Pre-therapeutic screening for Dihydropyrimidine dehydrogenase deficiency in measuring uracilemia in dialysis patients leads to a high rate of falsely positive results. Cancer Chemotherapy and Pharmacology, 88(6), pp.1049–1053. doi:https://doi.org/10.1007/s00280-021-04354-7.
Gamelin, E., Boisdron-Celle, M., Guérin-Meyer, V., Delva, R., Lortholary, A., Genevieve, F., Larra, F., Ifrah, N. and Robert, J. (1999). Correlation Between Uracil and Dihydrouracil Plasma Ratio, Fluorouracil (5-FU) Pharmacokinetic Parameters, and Tolerance in Patients With Advanced Colorectal Cancer: A Potential Interest for Predicting 5-FU Toxicity and Determining Optimal 5-FU Dosage. Journal of Clinical Oncology, 17(4), pp.1105–1105. doi:https://doi.org/10.1200/jco.1999.17.4.1105.
Gamelin, E., M. Boisdron-Celle, F Larra and Robert, J. (1997). A Simple Chromatographic Method for the Analysis of Pyrimidines and their Dihydrogenated Metabolites. Journal of Liquid Chromatography & Related Technologies, 20(19), pp.3155–3172. doi:https://doi.org/10.1080/10826079708000481.
García-González, X. et al. (2020). New DPYD variants causing DPD deficiency in patients treated with fluoropyrimidine. Cancer Chemotherapy and Pharmacology, 86(1), pp. 45–54. doi:10.1007/s00280-020-04093-1.
García-González, X. et al. (2016). ‘Clinical implementation of Pharmacogenetics’, Drug Metabolism and Personalized Therapy, 31(1). doi:10.1515/dmpt-2015-0031.
Gentile, G. et al. (2015). Genotype–phenotype correlations in 5-fluorouracil metabolism: A candidate DPYD haplotype to improve toxicity prediction. The Pharmacogenomics Journal, 16(4), pp. 320–325. doi:10.1038/tpj.2015.56.
Hariprakash, J. M., Vellarikkal, S. K., Keechilat, P., Verma, A., Jayarajan, R., Dixit, V., Ravi, R., Senthivel, V., Kumar, A., Sehgal, P., Sonakar, A. K., Ambawat, S., Giri, A. K., Philip, A., Sivadas, A., Faruq, M., Bharadwaj, D., Sivasubbu, S., & Scaria, V. (2018). Pharmacogenetic landscape of DPYD variants in south Asian populations by integration of genome-scale data. Pharmacogenomics, 19(3), 227-241. doi:10.2217/pgs-2017-0101.
Haute Autorité de Santé. (2018). Screening for dihydropyrimidine dehydrogenase deficiency to decrease the risk of severe toxicities related to fluoropyrimidines (5-fluorouracil or capecitabine) – INAHTA Brief. Saint-Denis La Plaine: HAS. [Accessed 18 Aug. 2023].
Henricks, L.M., Lunenburg, C.A.T.C., de Man, F.M., Meulendijks, D., Frederix, G.W.J., Kienhuis, E., Creemers, G.-J., Baars, A., Dezentjé, V.O., Imholz, A.L.T., Jeurissen, F.J.F., Portielje, J.E.A., Jansen, R.L.H., Hamberg, P., ten Tije, A.J., Droogendijk, H.J., Koopman, M., Nieboer, P., van de Poel, M.H.W. and Mandigers, C.M.P.W. (2018). DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. The Lancet Oncology, 19(11), pp.1459–1467. doi:https://doi.org/10.1016/s1470-2045(18)30686-7.
Henricks, L. M ., Opdam, F. L., Beijnen, J. H., Cats, A. & Schellens, J. H. M. (2017). DPYD genotype-guided dose individualization to improve patient safety of fluoropyrimidine therapy: call for a drug label update. Annals of Oncology. 28(12), 2915-2992. https://www.sciencedirect.com/science/article/pii/S0923753419353773#bb0135
Hishinuma, E., Gutiérrez Rico, E. and Hiratsuka, M. (2020). In Vitro Assessment of Fluoropyrimidine-Metabolizing Enzymes: Dihydropyrimidine Dehydrogenase, Dihydropyrimidinase, and β-Ureidopropionase. Journal of Clinical Medicine, 9(8), p.2342. doi:https://doi.org/10.3390/jcm9082342
Hodroj, K., Barthelemy, D., Lega, J.-C. ., Grenet, G., Gagnieu, M.-C. ., Walter, T., Guitton, J. and Payen-Gay, L. (2021). Issues and limitations of available biomarkers for fluoropyrimidine-based chemotherapy toxicity, a narrative review of the literature. ESMO Open, 6(3), p.100-125. doi:https://doi.org/10.1016/j.esmoop.2021.100125.
Innocenti, F., Mills, S. C., Sanoff, H., Ciccolini, J., Lenz, H. J. & Milano, G. (2020). All You Need to Know About DPYD Genetic Testing for Patients Treated With Fluorouracil and Capecitabine: A Practitioner-Friendly Guide. JCO Oncology Practice. 16(12), 793 – 798. https://ascopubs.org/doi/full/10.1200/OP.20.00553
Jennings, B.A. et al. (2012). Functional polymorphisms of folate metabolism and response to chemotherapy for colorectal cancer, a systematic review and meta-analysis. Pharmacogenetics and Genomics, 22(4), pp. 290–304. doi:10.1097/fpc.0b013e328351875d.
Jiang, H., Jiang, J., Hu, P. and Hu, Y. (2002). Measurement of endogenous uracil and dihydrouracil in plasma and urine of normal subjects by liquid chromatography–tandem mass spectrometry. Journal of Chromatography B, 769(1), pp.169–176. doi:https://doi.org/10.1016/s1570-0232(02)00009-0.
Kockum, I., Huang, J. & Stridh, P. (2023). Overview of Genotype Technologies and Methods. Current Protocols. 3(4). https://doi.org/10.1002/cpz1.727
Kristensen, M., Pedersen, P. and Mejer, J. (2010). The Value of Dihydrouracil/Uracil Plasma Ratios in Predicting 5-Fluorouracil-Related Toxicity in Colorectal Cancer Patients. Journal of International Medical Research, 38(4), pp.1313–1323. doi:https://doi.org/10.1177/147323001003800413.
Kuilenburg, van, Meinsma, R., Zonnenberg, B.A., Zoetekouw, L., Baas, F., Matsuda, K., Tamaki, N. and Gennip, van. (2003). Dihydropyrimidinase Deficiency and Severe 5-Fluorouracil Toxicity. Clinical Cancer Research, [online] 9(12), pp.4363–4367. https://aacrjournals.org/clincancerres/article/9/12/4363/203348/Dihydropyrimidinase-Deficiency-and-Severe-5
Laures, N., Konecki, C., Brugel, M., Giffard, A.-L., Abdelli, N., Botsen, D., Carlier, C., Gozalo, C., Feliu, C., Slimano, F., Djerada, Z. and Bouché, O. (2022). Impact of Guidelines Regarding Dihydropyrimidine Dehydrogenase (DPD) Deficiency Screening Using Uracil-Based Phenotyping on the Reduction of Severe Side Effect of 5-Fluorouracil-Based Chemotherapy: A Propension Score Analysis. Pharmaceutics, 14(10), p.2119. doi:https://doi.org/10.3390/pharmaceutics14102119.
Lu, Z., Zhang, R. and Diasio, R.B. (1993). Dihydropyrimidine dehydrogenase activity in human peripheral blood mononuclear cells and liver: population characteristics, newly identified deficient patients, and clinical implication in 5-fluorouracil chemotherapy. Cancer Research, 53(22), pp.5433–5438.
Lunenburg, C.A.T.C. et al. (2016). Prospective DPYD genotyping to reduce the risk of fluoropyrimidine-induced severe toxicity: Ready for prime time. European Journal of Cancer, 54, pp. 40–48. doi:10.1016/j.ejca.2015.11.008.
Maillard, M., Manon Launay, Royer, B., Jérôme Guitton, Elodie Gautier-Veyret, Broutin, S., Tron, C., Félicien Le Louedec, Ciccolini, J., Richard, D., Alarcan, H., Haufroid, V., Naïma Tafzi, Schmitt, A., Marie-Christine Etienne-Grimaldi, Céline Narjoz and Thomas, F. (2022). Quantitative impact of pre‐analytical process on plasma uracil when testing for dihydropyrimidine dehydrogenase deficiency. British Journal of Clinical Pharmacology, 89(2), pp.762–772. doi:https://doi.org/10.1111/bcp.15536.
Marin, C., Krache, A., Palmaro, C., Lucas, M., Hilaire, V., Ugdonne, R., De Victor, B., Quaranta, S., Solas, C., Lacarelle, B. and Ciccolini, J. (2020). A Simple and Rapid UPLC‐UV Method for Detecting DPD Deficiency in Patients With Cancer. Clinical and Translational Science, 13(4), pp.761–768. doi:https://doi.org/10.1111/cts.12762.
Martens, F. K., Huntjens, D. W., Rigter, T., Bartels, M., Bet P. M. & Cornel M. C. (2020). DPD Testing Before Treatment With Fluoropyrimidines in the Amsterdam UMCs: An Evaluation of Current Pharmacogenetic Practice. Frontiers in Pharmacology. https://doi.org/10.3389/fphar.2019.01609
Mercier, C., Brunet, C., Yang, C., Dupuis, C., Bagarry-Liegey, D., Duflo, S., Giovanni, A., Zanaret, M., Lacarelle, B., Duffaud, F. & Ciccolini, J. (2009). Pharmacoeconomic study in head and neck cancer patients: Impact of prospective DPD deficiency screening with 5-fluorouracil (5-FU) dose tailoring on toxicities-related costs. Journal of Clinical Oncology. 27(15). https://ascopubs.org/doi/abs/10.1200/jco.2009.27.15_suppl.6515
Meulendijks, D., Henricks, L.M., Jacobs, B.A.W., Aliev, A., Deenen, M.J., de Vries, N., Rosing, H., van Werkhoven, E., de Boer, A., Beijnen, J.H., Mandigers, C.M.P.W., Soesan, M., Cats, A. and Schellens, J.H.M. (2017). Pretreatment serum uracil concentration as a predictor of severe and fatal fluoropyrimidine-associated toxicity. British Journal of Cancer, 116(11), pp.1415–1424. doi:https://doi.org/10.1038/bjc.2017.94.
Milano, G. and Etienne, M.C. (1994). Dihydropyrimidine dehydrogenase (DPD) and clinical pharmacology of 5-fluorouracil (review). Anticancer Research, 14(6A), pp.2295–2297.
Milano, G. and Chamorey, A.-L. (2002). Clinical pharmacokinetics of 5-fluorouracil with consideration of chronopharmacokinetics. Chronobiology International, 19(1), pp.177–189. doi:https://doi.org/10.1081/cbi-120002597.
Ockeloen, C.W., Raaijmakers, A., Hijmans-van der Vegt, M., Bierau, J., de Vos-Geelen, J., Willemsen, A.E., van den Bosch, B.J. and Coenen, M.J. (2021). Potential added value of combined DPYD/DPD genotyping and phenotyping to prevent severe toxicity in patients with a DPYD variant and decreased dihydropyrimidine dehydrogenase enzyme activity. Journal of Oncology Pharmacy Practice, p.107815522110491. doi:https://doi.org/10.1177/10781552211049144.
Offer, S.M. et al. (2013). A DPYD variant (Y186C) in individuals of African ancestry is associated with reduced DPD enzyme activity. Clinical Pharmacology & Therapeutics, 94(1), pp. 158–166. doi:10.1038/clpt.2013.69.
Offer, S.M. et al. (2014). MicroRNAs mir-27a and Mir-27b directly regulate liver dihydropyrimidine dehydrogenase expression through two conserved binding sites. Molecular Cancer Therapeutics, 13(3), pp. 742–751. doi:10.1158/1535-7163.mct-13-0878.
Paulsen, N. H., Vojdeman, F., Andersen, A. E., Bergmann, T. K., Ewertz, M., Plomgaard, P., Hansen, M. R., Esbech, P. S., Pfeiffer, P., Qvortrup, C. & Damkier, P. (2022). DPYD genotyping and dihydropyrimidine dehydrogenase (DPD) phenotyping in clinical oncology. A clinically focused minireview. PubMed Central. 131(5), 325-346. https://doi.org/10.1111%2Fbcpt.13782
Paulsen, N.H., Vojdeman, F., Andersen, S.E., Bergmann, T.K., Ewertz, M., Plomgaard, P., Hansen, M.R., Esbech, P.S., Pfeiffer, P., Qvortrup, C. and Damkier, P. (2022). DPYD genotyping and dihydropyrimidine dehydrogenase (DPD) phenotyping in clinical oncology. A clinically focused minireview. Basic & Clinical Pharmacology & Toxicology, 131(5), pp.325–346. doi:https://doi.org/10.1111/bcpt.13782.
Robin, T., Saint-Marcoux, F., Toinon, D., Tafzi, N., Marquet, P. and El Balkhi, S. (2020). Automatic quantification of uracil and dihydrouracil in plasma. Journal of Chromatography B, 1142, p.122038. doi:https://doi.org/10.1016/j.jchromb.2020.122038.
Royer, B., Manon Launay, Ciccolini, J., Derain, L., François Parant, Thomas, F. and Jérôme Guitton. (2023). Impact of renal impairment on dihydropyrimidine dehydrogenase (DPD) phenotyping. ESMO open, 8(3), pp.101577–101577. doi:https://doi.org/10.1016/j.esmoop.2023.101577.
Salama, S., Gall0-Hershberg, D. & Forbes, L. (2023). Fluoropyrimidine Treatment in Patients with Dihydropyrimidine Dehydrogenase (DPD) Deficiency. Ontario Health Cancer Care Ontario.
Thomas, H.R., Ezzeldin, H.H., Guarcello, V., Mattison, L.K., Fridley, B.L. and Diasio, R.B. (2007). Genetic regulation of dihydropyrimidinase and its possible implication in altered uracil catabolism. Pharmacogenetics and Genomics, 17(11), pp.973–987. doi:https://doi.org/10.1097/fpc.0b013e3282f01788.
Thorn, C.F., Marsh, S., Carrillo, M. W., McLeond, H. L., Klein, T. E. & Altman, R. B. (2011). PharmGKB summary: fluoropyrimidine pathways. Available online: https://doi.org/10.1097%2FFPC.0b013e32833c6107 [Accessed 20/08/23]
Tong, C.C. et al. (2018). ‘A novel DPYD variant associated with severe toxicity of fluoropyrimidines: Role of pre-emptive DPYD genotype screening’, Frontiers in Oncology, 8. doi:10.3389/fonc.2018.00279.
White, C., Scott, R.J., Paul, C., Ziolkowski, A., Mossman, D. and Ackland, S. (2021). Ethnic Diversity of DPD Activity and the DPYD Gene: Review of the Literature. Pharmacogenomics and Personalized Medicine, 14, pp.1603–1617. doi:https://doi.org/10.2147/pgpm.s337147.
Yokoi, K. et al. (2020). Impact of DPYD, DPYS, and UPB1 gene variations on severe drug‐related toxicity in patients with cancer. Cancer Science, 111(9), pp. 3359–3366. doi:10.1111/cas.14553.
Yap, Y.-S. et al. (2017). ‘Predictors of hand-foot syndrome and pyridoxine for prevention of capecitabine–induced hand-foot syndrome’, JAMA Oncology, 3(11), p. 1538. doi:10.1001/jamaoncol.2017.1269.
Yoshino, T., Argilés, G., Oki, E., Martinelli, E., Taniguchi, H., Arnold, D., Mishima, S., Li, Y., Smruti, B.K., Ahn, J.B., Faud, I., Chee, C.E., Yeh, K.-H. ., Lin, P.-C. ., Chua, C., Hasbullah, H.H., Lee, M.A., Sharma, A., Sun, Y. and Curigliano, G. (2021). Pan-Asian adapted ESMO Clinical Practice Guidelines for the diagnosis, treatment and follow-up of patients with localised colon cancer. Annals of Oncology, 32(12), pp.1496–1510. doi:https://doi.org/10.1016/j.annonc.2021.08.1752.


{kind=link}