Abstract
Ageing leaves an epigenetic mark on every cell of the human body that changes the way some of the genes are expressed. These changes gradually accumulate to change and disrupt cellular identity, weaken repair systems and may lead to the development of cancer. This predictable drift has become so precise that it can be measured by using epigenetic clocks. These clocks are tools capable of estimating biological age with high accuracy. In this paper, we explore how errors in the age-related epigenomes cause gene mutations, stem-cell exhaustion and malignant transformations. We examine how biological age predicts the chance of getting cancer more reliably than chronological age. Finally, we investigate emerging technologies such as CRISPR-based epigenome editing that are beginning to reverse these molecular signs of ageing. This review shows that age-related epigenetic changes are not just simple indicators of decline but factors that increase a person’s likelihood of cancer development, and that targeted modification of these marks can reverse some age-related epigenetic changes. These findings suggest a future in which altering the epigenome may not only slow biological ageing but also reduce cancer risk before it begins.
Introduction
Cancer risk increases with age, and many scientists believe that this is not simply due to mutations. Ageing changes the epigenome by causing alterations in DNA methylation and histone modification patterns that control which genes are expressed. These changes can accumulate slowly and affect how cells function, especially in older tissues (Issa, 2014). Some of these changes are harmless but others may disrupt the cell’s ability to repair damage or control growth, which makes tumours easier to form (Baylin & Jones, 2016).
During ageing, DNA methylation patterns start to change across many tissues. Some regions lose methylation and others gain too much of it. These changes can limit the expression of important genes, including those that normally prevent damaged cells from growing (Horvath & Raj, 2018). Histone modifications also change with age, and this can affect how tightly DNA is packed. When these systems become unstable, the cell is not as efficient at repairing cells and damaged cells can build up more easily (Pal & Tyler, 2016). Over time, this may increase the risk of cells undergoing malignant transformations which can lead to the development of cancer. These predictable methylation changes are the reason epigenetic clocks exist. They measure specific sites in the genome to estimate biological age (Horvath & Raj, 2018). This suggests that the ageing epigenome has a direct link to why cancer appears more frequently in older adults.
New research is starting to test whether these age-related epigenetic changes can be slowed down or reversed. Early work with CRISPR editing, the delivery of epidrugs and reprogramming shows that some ageing marks can be reset (Fadul et al., 2023). If these methods become reliable, restoring a younger epigenetic pattern might lower cancer risk by improving cell repair and stability.
This paper looks at three ideas: (1) how age-related epigenetic drift contributes to errors in gene regulation and cancer; (2) how epigenetic clocks reflect cancer risk; and (3) how new tools may one day reset the epigenome to prevent age-related cancer.
Overview of Modification Pathways
Epigenetic machinery consists of DNA coiled with histones, forming nucleosomes. Each epigenetic mechanism is essential for normal development and maintenance of tissue-specific gene expression patterns in mammals (Sharma et al., 2010). These changes occur through epigenetic phenomena such as nucleosome remodelling, histone modifications, DNA methylation and miRNAs-mediated targeting of various genes. The nucleosome that chromatin is wrapped around consists of five types of histones. All five types of histones (H1, H2A, H2B, H3 and H4) can undergo post-transcriptional histone modifications (PTMs), which are positively charged and interact with negatively-charged DNA. These can involve acetylations and/or methylations, in which acetyl groups and methyl groups are attached to histones, or directly on DNA bases, by PTM (post-translational modification) writers called histone acetyltransferases (HATs) and DNA methyltransferases (DNMTs). These markers are read by PTM readers, which recruit transcriptional proteins to the DNA sequences. Hyperacetylation neutralises charges of lysine in histone proteins, weakening attraction to DNA, causing easier access of transcription proteins, which heightens gene expression; hypoacetylation compacts parts of the chromatin, reducing gene expression (Chen et al., 2023).
These epigenetic modifications are essential for normal mammalian development. Riggs (1975) tied DNA methylation to X-chromosome inactivation in females, an essential process preventing female cells from producing twice as many X-linked gene products as males. Bird (1986) then pioneered the characterisation of DNA methylation at CpG islands. His research found that methylated regions in cells occur as discrete islands – CpG islands, approximately 1-2 kbp long that occur every 100kbp. Bird found them to be either completely methylated or unmethylated near the promoter region of genes, suggesting their involvement in gene regulation and transcription. Histone modifications have been found to play a central role as well in transcription. Modification of histone tails primarily at lysine residues through methylation or acetylation alters the electrostatic interactions between the positively-charged histones and the negatively-charged phosphate groups in DNA (Dillinger, 2024). A decrease in the positive charge of the histone leads to loosely packed chromatin, known as euchromatin, which allows for transcription of DNA. However, an increase in the positive charge of the histones leads to tightly-packed chromatin, known as heterochromatin, which can silence certain genes.
Epigenetic Modifications Leading to Cancer
Cancer is a disease caused by many factors, but predominantly by modulation in gene expression. This causes complex networks ruling homeostasis to derange, allowing cells to mutate and grow without reference to the needs of our organism. The clear sets of cellular control pathways are ineffective or paralysed in almost all types of cancer (Hanahan & Weinberg, 2011). Proto-oncogens are genes that promote cell growth and, when mutated, become oncogenes, which lead to uncontrolled cell growth and cancer. Tumour suppressors also play a central role in regulating cell division and preventing uncontrolled cell growth, which can lead to cancer. Deregulated transcriptions of these genes, through activation of oncogenes and deactivation of tumour suppressors, have long been accepted to play a central role in carcinogenesis (Ilango et al., 2020).
Malignant cancer cells arise from regular cells through a multistep process involving both genetic and epigenetic changes. Similar to genetic lesions, epigenetic lesions are diverse in nature and serve to alter the structure and function of a genome, therefore participating in a cell’s acquisition of limitless uncontrolled growth and the phenotypic hallmark of the malignant cancer cell (Ilango et al., 2020). The degree of epigenetic difference between cancer cells and normal cells greatly exceeds the epigenetic differences seen between normal cells of different phenotypes. The complexity and frequency of epigenetic changes seen in cancer cells seem to defy explanations that rely on a single event (Futscher, 2012).
Alterations of DNA methylation can contribute to genomic instability and an increase in aneuploidies through faulty regulation of cell division, which are genetic conditions rising from an abnormal amount of chromosomes. Both of these are classic hallmarks of cancer (Hanahan & Weinberg, 2011). Figure 1 illustrates modifications to DNA involving DNA methylation which can lead to oncogenic phenotypes through several mechanisms. These include overall hypomethylation across the cancer genome, focal hypermethylation at tumour-suppressor gene promoters and mutagenesis of 5-methylcytosine-containing sequences by deamination, UV radiation or other carcinogenic exposures.
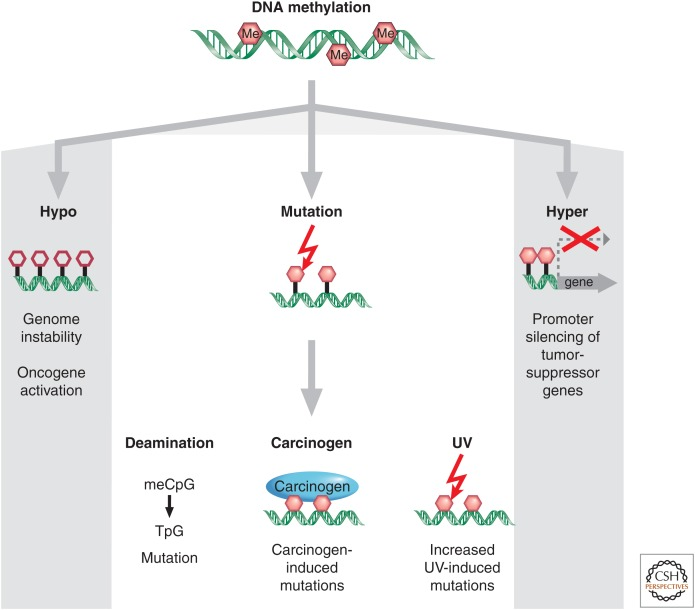
Figure 1: Pathways of DNA methylation in cancer (Baylin & Jones, 2016).
Effect of Ageing on Our Epigenome
Ageing is seen as an increase in metabolic dysfunctions throughout our body, being linked to adverse structural and functional changes in our organs. Over time, these losses in the efficiency of our physiological functions may increase susceptibility to disease and ultimately death. Harman (1992) postulates that ageing and its related diseases are caused by free radical-induced damage to cellular macromolecules and the inability to counterbalance the high levels of active oxygen molecules through endogenous anti-oxidant defences. Thus, ageing is strongly correlated to oxidative damage, structural modifications and loss of potency of several biomolecules (Maldonado et al., 2023). In relation to epigenetics, although there is no clear source of epigenetic damage, it is possible that ageing could trigger certain pathways, such as abnormal methylation, as many epigenetic changes are triggered by specific chemical environments. Low-level de novo methylation at CpG islands is known to occur in normal tissue and has been shown to increase with age (Issa, 2014).
DNA methylation and epigenetic inheritance show a significant change with age in mammals, most likely because of stochastic errors in epigenetic copying during stem cell replication. This drift results in gene expression mosaicism and has the potential to contribute to ageing phenotypes, including diseases (Issa, 2014). Figure 2 illustrates how stochastic errors in DNA methylation maintenance introduce epigenetic mosaicism in stem cell replication. Continued epigenetic mosaicism causes restricted differentiation in some stem cells, which leads to stem cell exhaustion and a selective growth advantage, all contributing to phenotypes and diseases of ageing.
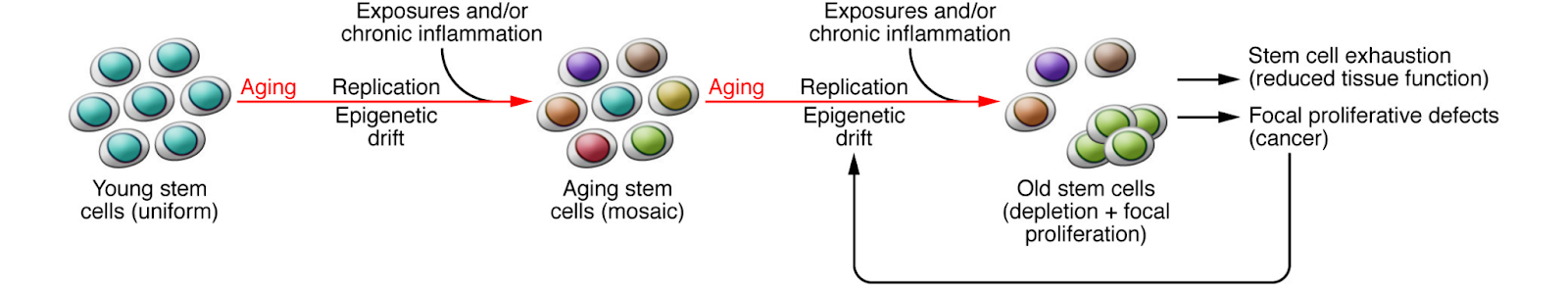
Figure 2: A model of the effects of the ageing epigenome on stem cell function (Issa, 2014).
Effect of an Ageing Epigenome on the Likelihood of Cancer
Accumulation of mutations is the common factor for ageing and cancer, and could be the explanation for why some diseases, such as cancer, increase exponentially rather than linearly with age. Oncogenic mutations accumulate with age in several tissues, such as blood, skin, the brain, oesophagus and liver. Sequencing studies on benign human nevi (moles), normal human skin and endometrium have revealed that oncogenic alterations are tolerated surprisingly well by normal adult human tissues and less well tolerated in aged tissue, leading to cancer (Martinocenara et al., 2015; Pollock et al., 2002; Moore,et al., 2020).
Whilst genome-wide levels of methylation decrease with age in all mammals, there is a tendency for DNA methylation to increase at specific sites, such as CpG islands, in the promoter regions of genes which are normally kept in an unmethylated state (Kim et al., 2005). The overall decrease in methylation levels is mirrored in tumour cells, a hallmark of all types of cancer, and is believed to contribute to carcinogenesis (Gaudet et al., 2003).
The unusual methylation of CpG islands is normally associated with stable and heritable gene silencing, such as chromosome X inaction in women. However, the hypermethylation and silencing of tumour suppressor genes is seen as an early event in many cancers, indicating that some DNA methylation patterns that occur during ageing might promote cancer (Feinberg et al., 2006). In two separate studies (Rakyan et al., 2010; Teschendorff et al., 2010), large-scale screens were performed for genes that are hypermethylated in ageing. They demonstrated significant overlap between these regions and the hypermethylated and silenced regions in cancer.
Younger Epigenomes vs Older Epigenomes
Epigenetic drift occurs when the epigenome starts to become less stable due to inaccurate DNA methylation. DNA methylation becomes less precise due to the many changes experienced as an individual ages (Zabransky et al., 2022), including the natural process of ageing, an individual’s diet and changing environments. Numerous discontinuous DNA methylation events, such as the ones previously mentioned, disrupt cellular function. Through the disruption of cellular function, hypomethylation and hypermethylation can later occur. This incorrect DNA methylation can indirectly cause cancer (Zabransky et al., 2022) through the silencing of the tumour suppressor genes – the genes that protect cells from cancers – and the activation of the oncogenes – the result of a mutation to a proto-gene (McCray, 2024). DNA methylation causes unnecessary changes resulting in further activation, causing a positive feedback system of hypomethylation. This hypomethylation, if intense and continuous enough, can create cancerous tumours through a process called tumourigenesis (McCray, 2024).
The average probability of developing cancer increases over the years as people age. In 2025, the average probability of cancer in individuals aged 50 and over was 20.4% in males and 16.4% in females (American Cancer Society, 2025); for younger individuals, the statistic was 3.4% in males and 5.9% in females (American Cancer Society, 2025). There are various reasons as to why younger epigenomes are more effective at cell and tissue repair; for example, the distinct gene structure. Younger genes are shorter and are less strongly expressed than older genes (Booth & Brunet, 2017). This is because the younger cells have high levels of various proteins like histone modifiers (able to change the structure of histones, the proteins that package DNA into nucleosomes) and chromatin modifiers (able to change the structure of chromatin, the complex of DNA and proteins that forms chromosomes inside the nucleus of eukaryotic cells). These modifications act like a switch to make DNA more accessible to transcriptions (Bannister & Kouzarides, 2011).
Existing Technologies in Epigenetics
Unlike the genome, the epigenome is flexible and dynamic. It can be heavily influenced by both external factors, such as diet, smoking and age, and altered in vivo. An example of environmental factors influencing histone acetylation is the condensing of acetyl groups at pro-inflammatory genes in response to cigarette smoke. Consequent loosening of chromatin structure leads to the overexpression of pro-inflammatory cytokines, leading to inflammatory conditions like COPD, bronchitis and asthma (Zong et al., 2019). These epigenetic modifications can be reversed and altered using epidrugs. The majority of epidrugs approved by the MHRA are HDAC inhibitors, which arrest the growth of – or completely destroy – cancer cells and DNMT inhibitors. Vorinostat, a HDAC inhibitor, inhibits the erasure of acetyl groups in hyperacetylated areas, which causes acetyl groups to condense, leading to cell cycle arrest/apostosis (Bubna, 2015). Vorinostat was found to suppress proteins essential for DNA repair such as RAD50 and MRE11, but only for cancer cells; normal cells were not affected. However, significant research is still to be made in the field, especially in terms of researching cytotoxicity (which Vorinostat displays in high amounts) and other limitations of epidrugs.
Furthermore, DNMT inhibitors such as Azacitidine prevent the methylation of DNA by methyltransferases, trapping DNMTs for the breakdown of the enzyme by the proteasome (proteasomal degradation) (Ilango et al., 2020). This demethylates CpG islands, leading to activation and promotion of tumour suppressor genes. However, many DNMT inhibitors are associated with side effects such as cytotoxicity, gastrointestinal issues and anaemia, among others. The hypomethylation of parts of the epigenome by DNMT inhibitors can also trigger the up-regulation of oncogenes like SALL4, which plays a large role in cancer progression and drug resistance. Figure 3 shows a drastic decrease in patient survival rate in the months after treatment with a hypomethylating agent such as Azacitidine (Liu et al., 2022) due to the promotion of SALL4 oncogenes.
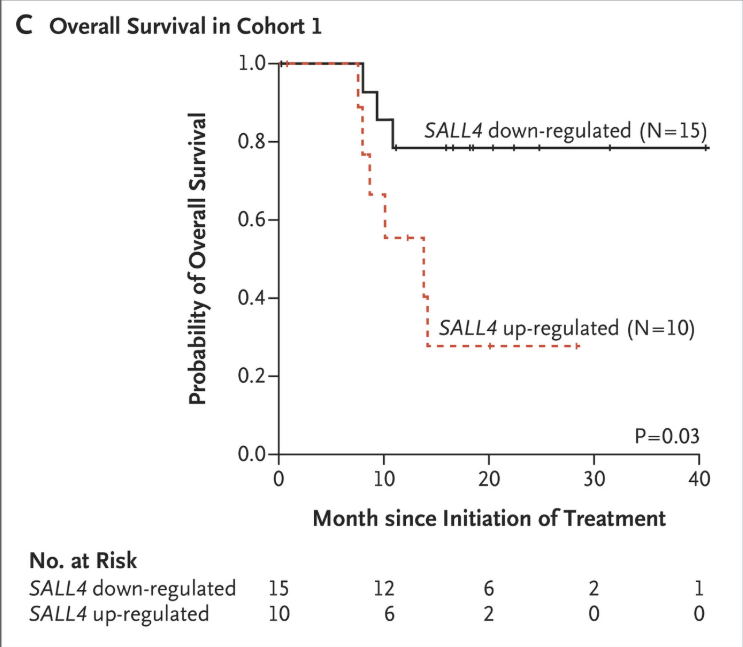
Figure 3: Patient survival rate in the months following treatment with DNMT inhibitors (Liu et al., 2022).
The delivery of epidrugs is heavily limited by their chemical properties that include a short half-life, poor solubility and permeability, and quick metabolic clearance. Therefore, epidrugs can be delivered using nano-particles, which have an ability to penetrate subcellular structures, release drugs over controlled periods of time (reducing rates of cytotoxicity) and preserve drugs against metabolic degradation (Yusuf et al., 2023). The use of prodrugs, which are chemically inactive drugs that can be activated at the target site, can temporarily change undesirable chemical properties such as those mentioned above (Suraweera et al., 2025). Furthermore, nanoformulations, specially designed, nanoparticle-based chemical formulas, can be used for delivery, such as liposomes and solid lipid nanoparticles (SLNs), which similarly provide chemical insolubility and resistance to metabolic degradation (Mukherjee et al., 2009). Without these more effective intra tumour deliveries, the effectiveness of epidrugs on solid tumours is hindered due to delivery limitations.
The use of modified CRISPR technology can also be used for highly specific and heritable epigenetic silencing and promotion. CRISPR-Cas9 is a gene editing tool exploited from natural defense mechanisms of bacterial cells to host invaders. The bacterial cell injects two short RNA strands, which can be customised by scientists to match the specific base sequence of the targeted gene section. These strands fuse to form a complex with a nuclease called Cas9, an enzyme capable of breaking down the phosphodiester bonds that hold the nucleotides together, essentially “cutting” the DNA (Redman et al., 2016). Once recognised by the DNA sequence of the organism, the targeted DNA complex “opens up”, allowing the RNA strands to join with the two halves of the DNA section, all within the Cas9-RNA complex. The new RNA prompts the creation of a double stranded break where the Cas9 protein severs the gene at two points, in which “desired genes” can be “edited” in. Desired genes can entail customised nucleotide sequences supplied by scientists for assimilation into the genome to produce desired effects.
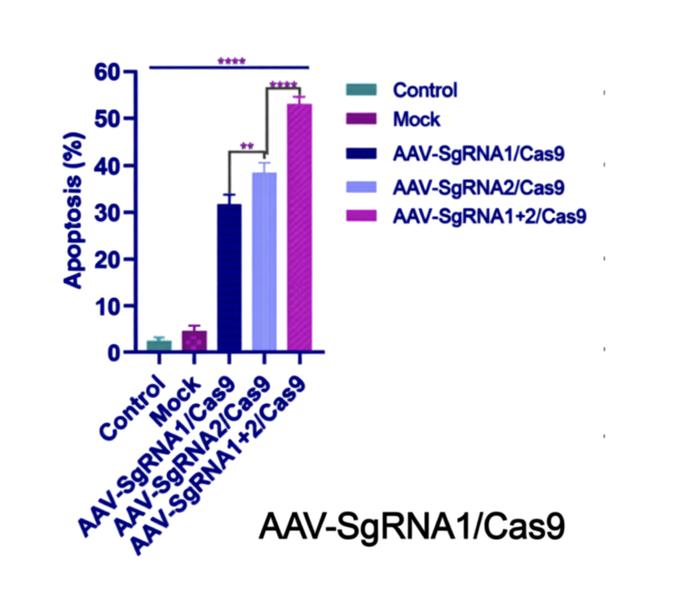
Figure 4: The effect of different CRISPR-Cas9 inducing cell apoptosis (x-axis) compared to control/mock groups without the CRISPR-Cas9 editing. The y-axis details the use of CRISPR-Cas9 complexes with different non-pathogenic virus vectors (AAVs) (Noroozi et al., 2022).
However, the nature of CRISPR-Cas9 editing means ultimately damaging the original genome. This can lead to cell apoptosis, as previously detailed, or the risk of drastic mutations while the cell tries to repair the double stranded break, including cancer. To avoid alterations to the genome itself, the accessibility of the genome can be changed, which is essentially the concept of epigenetics. CRISPR technology can use nuclease deactivated Cas9 (dCas9) proteins that can no longer cut DNA, but instead serve as a recruiter for DNA modification enzymes. dCas9s bind to target DNA sections, which can recruit HDAC and DNMT enzymes, which silence gene expression through removed acetyl groups and attached methyl tags. To improve the longevity and heritability of these epigenetic changes, a KRAB protein domain can be used in conjunction with the DMNT or HDACs (Nuñez et al., 2021). KRAB assigns repressor proteins such as KAP-1 during transcription to further silence the gene. This technique is the CRISPR-off technique, and is exponentially more heritable than CRISPR-Cas9 used alone. The CRISPR-Cas9 complex may also be fused with AAV vectors (adeno-associated viruses), a non-pathogenic delivery virus, due to their high tissue specificity, low immunogenicity and non pathogenic nature, forming gene editing tools such as AAV-SgRNA1/Cas9 (Naso et al., 2017).
Epigenetics are a driving factor in the ageing process, with older epigenetic profiles showing a global lack of heterochromatin (tightly packed chromatin as a result of acetylations and methylations) and overall DNA methylation. Rather than targeting specific gene expressions that result from an ageing epigenetic clock, the aged state of the epigenome itself can be reversed through the use of the Yamanaka factors, a set of four transcription factors that control gene expression (Pereira et al., 2024). Transcription factors are proteins that can bind directly to DNA to express or suppress a gene, much like DNMTs or HATs, except the Yamanaka factors ( Oct4, Sox2, Klf4 and c-Myc ) can remove epigenetic marks through cell reprogramming, restoring mature adult cells into a “youthful” epigenetic state where they can function without the hallmarks of ageing cells such as mitochondrial dysfunction, cellular senescence (cells in permanent growth arrest) and others. Crucially, this epigenetic clock can be reversed without the cells reaching a state of pluripotency, which would increase risk of tumourigenesis during cell differentiation (Pereira et al., 2024). Similar to CRISPR-Cas9 complexes, Yamanaka factors also used an adeno-associated virus delivery system, due to its high tissue specificity in targeting cells.
Ultimately, epigenetic therapy provides an alternative to other cancer treatments such as irreversible genetic alterations, which are prone to adverse effects and a promotion of oncogenes, or more commonly available options like immunotherapy, in which severe adverse events occur in around 20-60% of patients (Morad et al., 2021). It can also be used as a prognosis biomarker, in which, through comparing healthy epigenetic profiles that include DNA methylation and histone hyperacetylation to diseased ones, doctors can more easily assess whether a disease is high- or low-risk, plan future treatment and success rates that predict reactions to epidrugs, and even determine the risk of recurrence of cancers.
Conclusion
In summary, each epigenetic mechanism is essential for normal development and maintenance of tissue-specific gene expression patterns in mammals. These changes occur as part of epigenetic phenomena such as nucleosome remodelling, through histone modifications and DNA methylation mediated targeting of various genes. With increasing age, DNA methylation levels tend to decrease; however, there are many instances of hypermethylation of normally unmethylated CpG islands. These changes can lead to silencing of tumour-suppressor genes and activation of oncogenes. In comparison, younger epigenomes are generally more effective at repairing cells and tissues quickly and accurately. Our epigenetic clocks also play a central role in our predisposition to cancer. This is seen as epigenetic drifts along with epimutations which negatively impact the epigenome when an individual ages. These numerous mutations affect overall levels of DNA methylation, resulting in an increase in tumourigenesis. Epigenetics therapies represent a promising and significant avenue in cancer treatment, offering mechanisms that directly target the reversible modifications governing gene expressions. Although current epidrugs inhibitors demonstrate clear therapeutic potential, their undesirable chemical properties can be overcome through evolving technologies. As understanding of the epigenome continues to expand, epigenetic therapies and biomarkers may not only improve treatment outcomes but also enhance early diagnosis, risk assessment and personalised cancer care. Ultimately, continued advancements in epigenetics research and therapy have the potential to transform cancer care, prevention and future research.
Bibliography
American Cancer Society (2025) Cancer Facts & Figures 2025, CA: A Cancer Journal for Clinicians [online]. <https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2025/2025-cancer-facts-and-figures-acs.pdf>
Bannister, A. & Kouzarides, T. (2011) Regulation of Chromatin by Histone Modifications, Cell Research, 21, pp. 381-395.
Baylin, S.B. & Jones, P.A. (2016) Epigenetic Determinants of Cancer, Cold Spring Harb Perspect Biol, 8(9):a019505.
Bird, A. (1986) CpG-rich islands and the function of DNA methylation, Nature, 321, pp. 209–213.
Bubna A.K. (2015) Vorinostat-An Overview, Indian journal of dermatology, 60(4), p. 419.
Chen, Y.C., Koutelou, E. & Dent, S.Y.R. (2022) Now open: Evolving insights to the roles of lysine acetylation in chromatin organization and function, Mol Cell, 82(4), pp. 716-727.
Cleveland Clinic. (n.d.) Oncogenes, Cleveland Clinic [online]. <https://my.clevelandclinic.org/health/body/24949-oncogenes#additional-common-questions>
Dillinger, S. (2012). Epigenetics 101: Complete guide to understanding epigenetics, Active Motif [online]. <https://www.activemotif.com/epigenetics-101#summary>
Fadul S.M., Arshad, A. & Mehmood, R. (2023) CRISPR-based epigenome editing: mechanisms and applications, Epigenomics, 15(21), pp. 1137–1155.
Feinberg, A.P., Ohlsson, R. & Henikoff, S. (2006) The epigenetic progenitor origin of human cancer, Nat Rev Genet, 7(1):21-33.
Futscher, B.W. (2012) Epigenetic changes during cell transformation, Epigenetic Alterations in Oncogenesis, pp. 179-194.
Gaudet, F., Hodgson, J.G., Eden, A., Jackson-Grusby, L., Dausman, J., Gray, J.W., Leonhardt, H. & Jaenisch, R. (2003) Induction of tumours in mice by genomic hypomethylation, Science, 300(5618):489-92.
Hanahan, D. & Weinberg, R.A. (2011) Hallmarks of cancer: the next generation, Cell, 144(5), pp. 646-74.
Harman, D. (1992) Free radical theory of ageing, Mutation Research, 275(3-6), pp. 257-266.
Horvath, S. & Raj, K. (2018) DNA methylation-based biomarkers and the epigenetic clock theory of ageing’, Nature Reviews Genetics, 19, pp. 371–384.
Ilango, S., Paital, B., Jayachandran, P., Padma, P.R. & Nirmaladevi, R. (2020) Epigenetic alterations in cancer, Front. Biosci. (Landmark Ed), 25(6), pp. 1058–1109.
Issa, J.P. (2014) Ageing and epigenetic drift: a vicious cycle, The Journal of clinical investigation, 124(1), pp. 24–29.
Kim, J.Y., Tavaré, S. & Shibata, D. (2005) Counting human somatic cell replications: methylation mirrors endometrial stem cell divisions, Proc Natl Acad Sci USA, 102(49):17739-44.
Liu, Y.C., Kwon, J., Fabiani, E., Xiao, Z., Liu, Y.V., Follo, M.Y. et al. (2022) Demethylation and Up-Regulation of an Oncogene after Hypomethylating Therapy, The New England journal of medicine, 386(21), pp. 1998–2010.
Maldonado, E., Morales-Pison, S., Urbina, F. & Solari, A. (2023) Ageing Hallmarks and the Role of Oxidative Stress, Antioxidants (Basel), 2(3):651.
Martincorena, I., Roshan, A., Gerstung, M., Ellis, P., Van Loo, P., McLaren, S. et al. (2015) Tumour Evolution: High burden and pervasive positive selection of somatic mutations in normal human skin, Science, 348(6237):880-6.
McCray A. (2024) Epigenetics and Cancer Treatment, WebMD [online]. <https://www.webmd.com/cancer/cancer-treatment-epigenetics>
Meiers, I. (2010) Glutathione S-Transferase pi (GSTP1), Atlas Genet Cytogenet Oncol Haematol, 14(12), pp.1181 – 1185.
Moore, L., Leongamornlert, D., Coorens, T.H.H., Sanders, M.A., Ellis, P. et al. (2020) The mutational landscape of normal human endometrial epithelium, Nature, 580(7805):640-646.
Morad, G., Helmink, B.A, Sharma, P. & Wargo, J.A. (2021) Hallmarks of response, resistance, and toxicity to immune checkpoint blockade, A Cell Press Journal, 184(21), pp. 5309-5337.
Mukherjee, S., Ray, S., & Thakur, R.S. (2009) Solid lipid nanoparticles: a modern formulation approach in drug delivery system, Indian journal of pharmaceutical sciences, 71(4), pp. 349–358.
Naso, M.F., Tomkowicz, B., Perry, W.L. & Strohl, W.R. (2017) Adeno-Associated Virus (AAV) as a Vector for Gene Therapy, BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy, 31(4), pp. 317–334.
Noroozi, Z., Shamsara, M., Valipour, E., Esfandyari, S., Ehghaghi, A. et al. (2022). Antiproliferative effects of AAV-delivered CRISPR/Cas9-based degradation of the HPV18-E6 gene in HeLa cells, Scientific Reports, 12.
Nuñez, J.K., Chen, J., Pommier, G.C., Cogan, J.Z., Replogle, J.M., Adriaens, C. et al. (2021) Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing, Cell, 184(9):e17, pp. 2503–2519.
Pal, S. & Tyler, J.K. (2016) Epigenetics and Ageing, Science Advances, 2(7):e1600584.
Pereira, B., Correia, F.P., Alves, I.A., Costa, M., Gameiro, M., Martins, A.P. & Saraiva, J.A. (2024) Epigenetic reprogramming as a key to reverse ageing and increase longevity, Ageing research reviews, 95, 102204.
Pollock, P.M., Harper, U.L., Hansen, K.S., Yudt, L.M., Stark, M. et al. (2003) High frequency of BRAF mutations in nevi, Nat Genet, 33(1):19-20.
Rakyan, V.K., Down, T.A., Maslau, S., Andrew, T., Yang, T.P. et al. (2010) Human Ageing-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains, Genome Res, 20(4):434-9.
Redman, M., King, A., Watson, C. & King, D. (2016) What is CRISPR/Cas9?, Archives of disease in childhood. Education and practice edition, 101(4), pp. 213–215.
Riggs, A.D. (1975) X inactivation, differentiation, and DNA methylation, Cytogenet Cell Genet, 14(1), pp. 9-25.
Suraweera, A., O’Byrne, K.J. & Richard, D.J. (2025) Epigenetic drugs in cancer therapy, Cancer Metastasis Rev, 44(37).
Teschendorff, A.E., Menon, U., Gentry-Maharaj, A., Ramus, S.J., Weisenberger, D.J., Shen, H. et al. (2010) Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer, Genome Res, 20(4):440-6.
Yusuf, A., Almotairy, A.R.Z., Henidi, H., Alshehri, O.Y. & Aldughaim, M.S. (2023) Nanoparticles as Drug Delivery Systems: A Review of the Implication of Nanoparticles’ Physicochemical Properties on Responses in Biological Systems, Polymers, 15(7), 1596.
Zabransky, D.J., Jaffee, E.M. & Weeraratna, A.T. (2023) Shared genetic and epigenetic changes link Ageing and cancer, Trends Cell Biol, 32(4), pp. 338-350.
Zong, D., Liu, X., Li, J., Ouyang, R. & Chen, P. (2019) The role of cigarette smoke-induced epigenetic alterations in inflammation, Epigenetics & Chromatin, 12(1), p. 65.


{kind=link}